
{kind=link}
[ad_1]
With the discharge of platforms like DALL-E 2 and Midjourney, diffusion generative fashions have achieved mainstream reputation, owing to their means to generate a collection of absurd, breathtaking, and infrequently meme-worthy photographs from textual content prompts like “teddy bears working on new AI research on the moon in the 1980s.” But a crew of researchers at MIT’s Abdul Latif Jameel Clinic for Machine Learning in Health (Jameel Clinic) thinks there could possibly be extra to diffusion generative fashions than simply creating surreal photographs — they might speed up the event of recent medication and cut back the chance of hostile negative effects.
A paper introducing this new molecular docking mannequin, known as DiffDock, will likely be offered on the eleventh International Conference on Learning Representations. The mannequin’s distinctive method to computational drug design is a paradigm shift from present state-of-the-art instruments that almost all pharmaceutical corporations use, presenting a serious alternative for an overhaul of the normal drug improvement pipeline.
Drugs usually operate by interacting with the proteins that make up our our bodies, or proteins of micro organism and viruses. Molecular docking was developed to realize perception into these interactions by predicting the atomic 3D coordinates with which a ligand (i.e., drug molecule) and protein might bind collectively.
While molecular docking has led to the profitable identification of medicine that now deal with HIV and most cancers, with every drug averaging a decade of improvement time and 90 p.c of drug candidates failing pricey scientific trials (most research estimate common drug improvement prices to be round $1 billion to over $2 billion per drug), it’s no surprise that researchers are in search of sooner, extra environment friendly methods to sift via potential drug molecules.
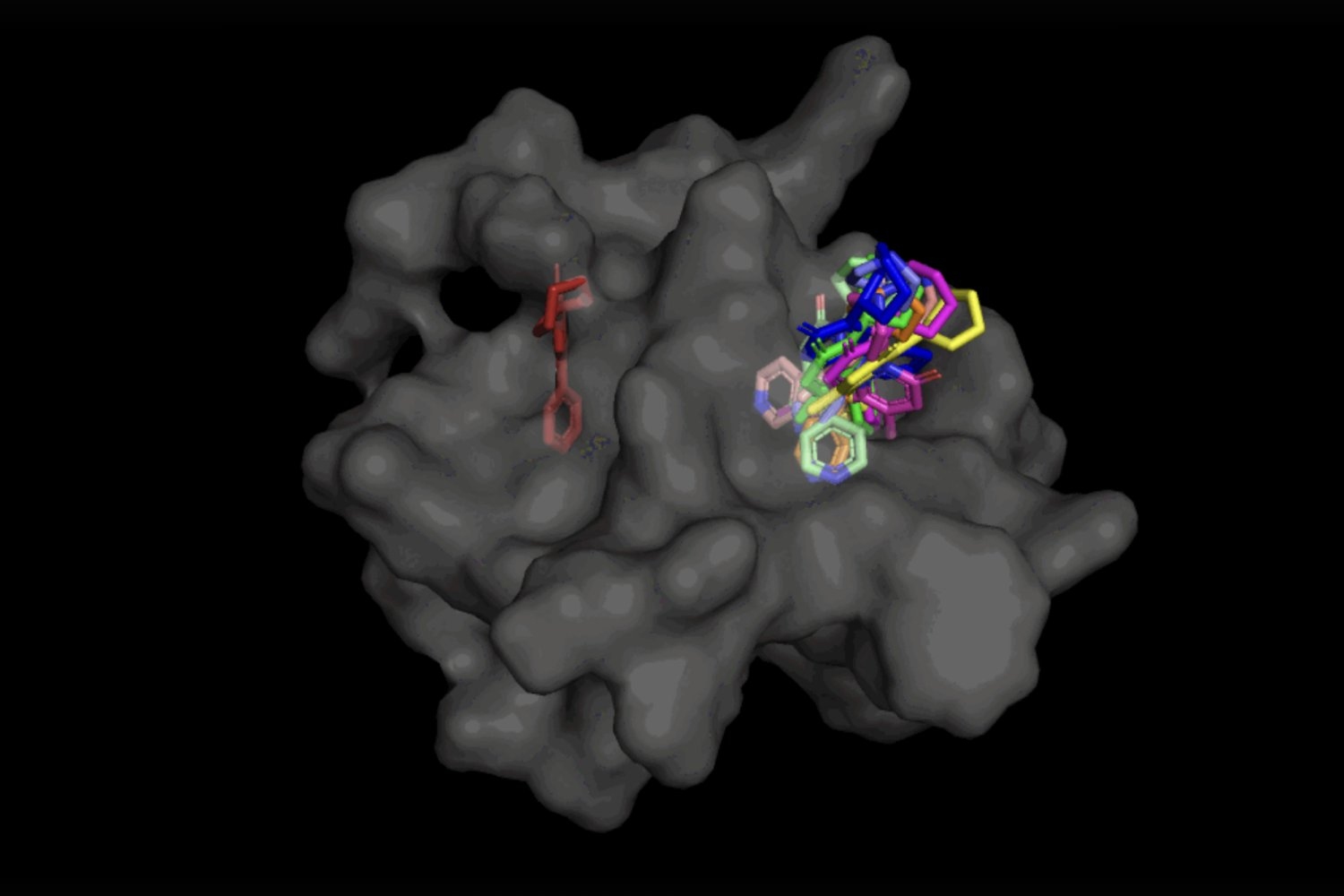
Currently, most molecular docking instruments used for in-silico drug design take a “sampling and scoring” method, trying to find a ligand “pose” that most closely fits the protein pocket. This time-consuming course of evaluates numerous completely different poses, then scores them based mostly on how effectively the ligand binds to the protein.
In earlier deep-learning options, molecular docking is handled as a regression drawback. In different phrases, “it assumes that you have a single target that you’re trying to optimize for and there’s a single right answer,” says Gabriele Corso, co-author and second-year MIT PhD pupil in electrical engineering and pc science who’s an affiliate of the MIT Computer Sciences and Artificial Intelligence Laboratory (CSAIL). “With generative modeling, you assume that there is a distribution of possible answers — this is critical in the presence of uncertainty.”
“Instead of a single prediction as previously, you now allow multiple poses to be predicted, and each one with a different probability,” provides Hannes Stärk, co-author and first-year MIT PhD pupil in electrical engineering and pc science who’s an affiliate of the MIT Computer Sciences and Artificial Intelligence Laboratory (CSAIL). As a consequence, the mannequin does not have to compromise in trying to reach at a single conclusion, which is usually a recipe for failure.
To perceive how diffusion generative fashions work, it’s useful to elucidate them based mostly on image-generating diffusion fashions. Here, diffusion fashions steadily add random noise to a 2D picture via a collection of steps, destroying the information within the picture till it turns into nothing however grainy static. A neural community is then educated to get better the unique picture by reversing this noising course of. The mannequin can then generate new knowledge by ranging from a random configuration and iteratively eradicating the noise.
In the case of DiffDock, after being educated on quite a lot of ligand and protein poses, the mannequin is ready to efficiently establish a number of binding websites on proteins that it has by no means encountered earlier than. Instead of producing new picture knowledge, it generates new 3D coordinates that assist the ligand discover potential angles that might permit it to suit into the protein pocket.
This “blind docking” method creates new alternatives to benefit from AlphaFold 2 (2020), DeepMind’s well-known protein folding AI mannequin. Since AlphaFold 1’s preliminary launch in 2018, there was a substantial amount of pleasure within the analysis group over the potential of AlphaFold’s computationally folded protein constructions to assist establish new drug mechanisms of motion. But state-of-the-art molecular docking instruments have but to reveal that their efficiency in binding ligands to computationally predicted constructions is any higher than random probability.
Not solely is DiffDock considerably extra correct than earlier approaches to conventional docking benchmarks, because of its means to cause at the next scale and implicitly mannequin a few of the protein flexibility, DiffDock maintains excessive efficiency, at the same time as different docking fashions start to fail. In the extra sensible state of affairs involving using computationally generated unbound protein constructions, DiffDock locations 22 p.c of its predictions inside 2 angstroms (broadly thought of to be the edge for an correct pose, 1Å corresponds to at least one over 10 billion meters), greater than double different docking fashions barely hovering over 10 p.c for some and dropping as little as 1.7 p.c.
These enhancements create a brand new panorama of alternatives for organic analysis and drug discovery. For occasion, many medication are discovered by way of a course of often known as phenotypic screening, through which researchers observe the consequences of a given drug on a illness with out figuring out which proteins the drug is appearing upon. Discovering the mechanism of motion of the drug is then vital to understanding how the drug will be improved and its potential negative effects. This course of, often known as “reverse screening,” will be extraordinarily difficult and expensive, however a mix of protein folding methods and DiffDock might permit performing a big a part of the method in silico, permitting potential “off-target” negative effects to be recognized early on earlier than scientific trials happen.
“DiffDock makes drug target identification much more possible. Before, one had to do laborious and costly experiments (months to years) with each protein to define the drug docking. But now, one can screen many proteins and do the triaging virtually in a day,” Tim Peterson, an assistant professor on the University of Washington St. Louis School of Medicine, says. Peterson used DiffDock to characterize the mechanism of motion of a novel drug candidate treating aging-related ailments in a latest paper. “There is a very ‘fate loves irony’ aspect that Eroom’s law — that drug discovery takes longer and costs more money each year — is being solved by its namesake Moore’s law — that computers get faster and cheaper each year — using tools such as DiffDock.”
This work was carried out by MIT PhD college students Gabriele Corso, Hannes Stärk, and Bowen Jing, and their advisors, Professor Regina Barzilay and Professor Tommi Jaakkola, and was supported by the Machine Learning for Pharmaceutical Discovery and Synthesis consortium, the Jameel Clinic, the DTRA Discovery of Medical Countermeasures Against New and Emerging Threats program, the DARPA Accelerated Molecular Discovery program, the Sanofi Computational Antibody Design grant, and a Department of Energy Computational Science Graduate Fellowship.
[ad_2]